ノンターゲットメタボロミクス解析ソフトウェア
Progenesis QI

ダウンロードサイトより、Progenesis QI for metabolomicsをダウンロードして、実際に使用していただくことができます。
ダウンロードページ(メーカーサイト)
CE-MSから取得されたメタボロミクスデータの比較定量解析を行うソフトウェアです。
高精度アライメントにより、測定間のリテンションタイムのズレを精確に補正し、ピーク強度からデータ間の相対定量を行います。
比較定量だけでなく、付加イオンのデコンボリューション~同定~多変量解析までひとつのソフトウェアでシームレスに対応できます。
データを視覚的に見やすく可視化できるため理解が容易で、メタボロミクス初心者の方にもお奨めのソフトウェアです。
ワークフローが簡単!
ソフトウェアの指示に従っていただくだけで簡単に解析が可能です。
2Dマップ表示でアラインメントが精確&簡単!
ピクセルレベルでの精細なアライメントが可能です。2Dマップ表示されるためアライメントの様子を見やすく可視化できます。

欠損値がない!
欠損値が生じないため、最も大きな変化を正しく定量することができます。
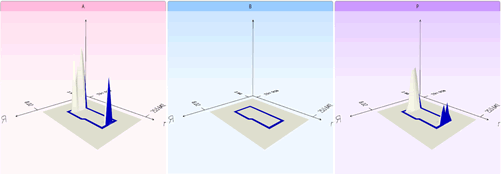
付加イオンのデコンボリューション
同じメタボライトに由来する付加イオンをまとめることができます。
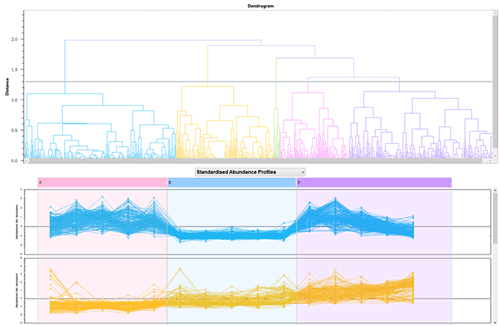
多変量解析
主成分分析(PCA)やクラスター解析などの多変量解析をシームレスに行うことが可能です。 欠損値が生じないのですべてのピークの多変量解析が可能です。
製品開発の背景
プロテオームのショットガン解析などのLC-MS解析データには多数のペプチド情報が含まれておりますが、LC-MS/MSの性能的限界のために全てのペプチドを同定することはできません。 LC-MSを用いた従来の発現差異解析法では、同定できなかったペプチドは解析対象にならず、比較的イオン強度の高い一部のペプチドのみが解析されてきました。 特に安定同位体標識を用いた解析ではMS/MSデータ上で定量比較が行われるため、MS/MSデータの存在しないペプチドピークは同定も比較もされません。
LC-MSデータでの比較定量
~MS/MSデータに依存しない、網羅的比較解析が可能~
Progenesis QI for proteomicsは、LC-MSデータ上のピークボリュームをサンプル間で比較するため、MS/MSデータの有無にかかわらず、LC-MSデータ上の全ピークを定量比較することができます。 特にバイオマーカー探索においては、多くのピークを比較解析することが重要です。 多くのピークを比較解析することで、優れたマーカー候補を発見できる可能性を飛躍的に向上させています。
定量比較解析を正確に行うため、Progenesis QI for proteomicsには下記の機能が搭載されています。
リテンションタイム補正機能:トータルイオンクロマトグラムでの補正ではなく、ピークごとに補正します。
ピークボリューム積算機能: イオン強度を積算してボリューム(体積)として認識しますので、定量性が確保されます。
ノーマライゼーション機能: イオンスプレイの安定性を考慮し、各測定間の標準化を行います。
ノイズ除去機能: アイソトープイオンが検出できないピークをノイズとして除去します。
フィルタリング機能: イオン価数を指定してフィルターをかけることができるため、ペプチド以外のピークを除去できます。
マージ機能: 各LC-MSデータを統合してからピーク検出を行うため、欠損値が生じません。
比較定量後の同定
~興味あるピークを集中的に同定できます~
Progenesis QI for proteomicsでは、LC-MSデータもしくはLC-MS/MSデータを解析することができます。 LC-MS/MSデータを用いて比較解析を行った場合、ピークエリア内にMS/MSデータが存在する場合には、ペプチド同定結果を表示することができます(別途MASCOTなどMS/MSイオンサーチエンジンが必要です)。
MS/MSデータが存在しない場合、あるいは同定スコアが十分ではないピークのインクルージョンリストを作成することができます(アプライドバイオシステムズ、サーモ、ウォーターズなど各社LC-MS/MS装置に対応)。 このインクルージョンリストを用いてLC-MS/MS測定することで、未同定のピークを中心に同定作業を進めることができます。 さらに、有意差検定、多変量解析で興味のあるピークを絞り込んでインクルージョンリストを作成することもできます。興味のあるピークを集中的に同定することで、貴重なサンプルを無駄にすることがありません。
バイオマーカー探索では、疾患群とコントロール群での有意差が高いピークのうち、未同定ピークをインクルージョンリストとして出力し、集中的に同定作業を進めることが有効です。
Progenesis QI for proteomicsでは、翻訳後修飾部位を含むペプチドの解析や、タンパク質レベルでの差異解析にも対応しています。
製品/シリーズに関するお問い合わせ
製品に関するご質問や技術的なお問い合わせなどはこちらのフォームをご利用ください。